
La miastenia gravis: una review aggiornata del Prof. Gandini
In questo nuovo blog il nostro esperto in neurologia, Prof. Gandini dell'Università di Bologna, ci guida alla scoperta di questa affascinante patologia neuro-muscolare.
PER COMPRENDERE MEGLIO: UN RIPASSO DELL’ANATOMIA FUNZIONALE
Il Sistema Nervoso Periferico (SNP) si compone di una parte efferente, con funzioni motorie, e di una parte afferente, con funzione sensitiva.
La componente efferente (motoria) del SNP viene nella pratica clinica identificata con il sistema neuromuscolare (o sistema del motoneurone inferiore) che, come indicato dal nome, include anche il muscolo costituendo un’unità morfo-funzionale e clinica indissolubile.
L’unità anatomofunzionale del sistema neuromuscolare è l’unità motoria. Il sistema neuromuscolare è infatti costituito da molteplici unità motorie, ognuna delle quali si compone di una cellula nervosa (l’α-motoneurone), della giunzione neuromuscolare e di tutte le fibre muscolari direttamente innervate da quel motoneurone.
L’identificazione della componente efferente del SNP con il sistema neuromuscolare è impropria, in quanto una parte di quest’ultimo è anatomicamente localizzata nel SNC. L’α-motoneurone è infatti un particolare tipo di neurone efferente, il cui corpo cellulare è localizzato nella sostanza grigia delle corna ventrali del midollo spinale, cioè nel SNC. L’assone dell’α-motoneurone lascia il midollo spinale come parte della radice ventrale ed entra nella costituzione del nervo spinale, nel quale decorre fino ad arrivare a contrarre sinapsi a livello dei muscoli come parte di uno specifico nervo periferico, quest’ultimo costituito dal contributo di più nervi spinali.
In prossimità del muscolo l’assone dell’α-motoneurone si ramifica in modo proporzionale alle fibre muscolari che va ad innervare. L’unità motoria che viene a formarsi è costituita da un numero variabile di fibre muscolari, in dipendenza del tipo di attività che il motoneurone presiede.
La giunzione neuromuscolare (GNM) è il punto di contatto tra la parte terminale dell’assone e il muscolo scheletrico e svolge un ruolo di fondamentale importanza nella contrazione muscolare.
La giunzione neuromuscolare può essere suddivisa in tre componenti di base:
- La membrana presinaptica, appartenente all’assone
- Lo spazio intersinaptico
- La membrana postsinaptica, appartenente al muscolo scheletrico.
Il neurotrasmettitore liberato a livello di GNM è l’Acetilcolina (ACh). Affinché la contrazione muscolare possa avvenire, il potenziale d’azione dell’assone deve essere trasmesso al muscolo dalla giunzione GNM. Il potenziale d’azione arriva sul bottone terminale dell’assone (membrana presinaptica) provocando la depolarizzazione di quest’area e la conseguente apertura dei canali del Calcio, permettendo l’entrata di questo ione all’interno del terminale presinaptico. L'aumentata concentrazione intracellulare di ioni Calcio innesca la fusione delle vescicole che contengono l’ACh con la membrana cellulare permettendo l'esocitosi dell’ACh nello spazio intersinaptico.
Le molecole di ACh attraversano lo spazio intersinaptico per raggiungere specifici recettori, situati sulla membrana post-sinaptica del muscolo. Questi recettori, costituiti da 5 subunità proteiche, fungono da canali transmembranari per il Sodio. Il legame tra l’ACh e il recettore modifica la conformazione del recettore che, aprendosi, permette l’entrata di ioni Sodio a livello della fibrocellula muscolare, provocando così una depolarizzazione locale denominata potenziale di placca (EPP: End Plate Potential).
L’EPP deve raggiungere una certa soglia sommandosi ad altri potenziali simili per arrivare a produrre la depolarizzazione del muscolo e la conseguente contrazione. L'entità dell'EPP dipende infatti dal numero di recettori per l’ACh attivati. Nel soggetto normale, esiste una sovrabbondanza sia di ACh che di recettori per l’ACh disponibili e, pertanto, il potenziale prodotto è estremamente superiore a quanto richiesto per la contrazione muscolare. Questo eccesso è chiamato il fattore di sicurezza (Safety Factor) della trasmissione neuromuscolare.
Durante la prosecuzione del movimento si ha una depolarizzazione ripetuta del terminale assonico e la quantità di ACh liberata nello spazio intersinaptico gradualmente, ad ogni evento depolarizzante, diminuisce. Il fenomeno, assolutamente normale, è chiamato Rundown.
Nei soggetti normali, gli effetti del rundown sono trascurabili per via dell’ampio Safety Factor presente. Il rundown può invece diventare significativo in quei soggetti (come quelli miastenici) in cui il safety factor (in questo caso la sovrabbondanza di recettori per l’ACh disponibili) diminuisca sensibilmente, producendo una “debolezza” che diventa clinicamente rilevante.
Va sottolineato che a volte è difficile, da un punto di vista clinico, differenziare tra un problema a carico selettivamente del nervo, del muscolo o della giunzione neuromuscolare. Dopo la visita ci si limita pertanto a definire il complesso dei sintomi caratteristici come “sindrome neuromuscolare” (o “sindrome del motoneurone inferiore”).
LA MIASTENIA GRAVIS
La miastenia gravis (MG) può presentarsi sia come malattia congenita che come malattia acquisita.
Le sindromi miasteniche congenite sono un gruppo di malattie ereditarie caratterizzate da disturbi della trasmissione a livello di giunzione neuromuscolare che possono coinvolgere le regioni pre- e post sinaptiche della GNM.
Negli ultimi anni diversi geni sono stati scoperti come fattori causali della malattia in diverse razze di cani, tra cui il Jack Russell Terrier, nell’Old Danish Pointing dog, nel Labrador retriever e nel bassotto. Tipicamente, i segni clinici compaiono allo svezzamento e possono interessare più soggetti di una stessa cucciolata.
La miastenia gravis acquisita è uno degli esempi più studiati e compresi di malattia autoimmune. Nel cane, la malattia è data dalla presenza di anticorpi umorali contro i recettori per l’ACh. L’attacco degli anticorpi e la successiva lisi mediata dal complemento produce una riduzione del numero dei recettori per l’ACh a livello di membrana postsinaptica. Per questo motivo, nel soggetto con Miastenia gravis acquisita il margine del Safety Factor diventa estremamente ridotto, per cui stimolazioni ripetute del nervo producono un rundown che diventa clinicamente significativo. È questa la patogenesi dell’intolleranza all’esercizio che caratterizza la malattia.
Nella forma autoimmune, l’origine degli anticorpi rimane sconosciuta. Esiste però la possibilità che la malattia sia associata a neoplasie, come il timoma e, meno frequentemente, il linfoma cutaneo e altre neoplasie; in questi casi la miastenia entra nel contesto di una sindrome paraneoplastica a meccanismo immunomediato.
PRESENTAZIONE CLINICA
La miastenia gravis acquisita è stata osservata soprattutto nei cani di taglia medio-grande e, per quanto riguarda la razza, soprattutto nel Pastore tedesco, nel Golden retriever e nel Labrador. L’età di insorgenza è bimodale, nel senso che i soggetti manifestano la malattia principalmente o sotto i 5 anni o sopra i 7 anni. I gatti manifestano la malattia soprattutto tra i 2-3 e, in seguito, tra i 9 e 10 anni.
In termini di presentazione clinica, si possono distinguere tre forme cliniche distinte di miastenia: focale, generalizzata e fulminante.
La MG focale si presenta con debolezza di un gruppo limitato di muscoli, come quelli dell’esofago o i muscoli laringei, faringei e facciali. I segni clinici sono nella maggior parte dei casi riconducibili alla presenza di megaesofago e comprendono rigurgito, disfagia, atti di deglutizione a vuoto e tosse. Può essere presente inoltre disfonia. L’esame neurologico può evidenziare la diminuzione del riflesso della deglutizione e palpebrale, nonché un ridotto tono della mandibola. Studi retrospettivi descrivono come colpiti dalla forma focale il 36-43% dei cani e il 15% dei gatti.
E’ verosimile pensare che diversi casi considerati in passato come megaesofago idiopatico fossero in realtà casi di MG focale. In un recente studio (2020) su 99 cani, i casi di megaesofago idiopatico erano il 42.7% mentre quelli associati a MG focale erano il 38.2%.
La MG generalizzata è caratterizzata da progressiva debolezza, facile affaticamento, tremori durante la stazione e intolleranza all’esercizio che si manifesta con soste frequenti durante il movimento e con incapacità progressiva a continuare la deambulazione. Dopo il riposo i soggetti sono in grado di camminare normalmente per un periodo variabilmente breve prima di ricominciare ad affaticarsi nuovamente. La debolezza generalizzata è riportata nel 57-64% dei cani e nell’80% dei gatti affetti da MG. Nel gatto è abbastanza frequente osservare, come segno di grave debolezza, la ventroflessione del collo.
Se il soggetto ha avuto la possibilità di riposarsi per un tempo adeguato, l’esame neurologico è assolutamente normale. Anche nella forma generalizzata sono a volte presenti i deficit dei nervi cranici descritti per la forma focale. Ai segni locomotori è spesso associato il rigurgito per la presenza di megaesofago. La broncopolmonite ab ingestis, testimoniata da stress respiratorio e tosse profonda, è la più frequente e temuta complicazione.
La MG fulminante è una forma non frequente e gravissima di miastenia generalizzata iperacuta, caratterizzata dal grave coinvolgimento dei muscoli appendicolari e soprattutto respiratori, che porta l’animale al decubito e ad una respirazione estremamente difficoltosa che può esitare rapidamente nel decesso del soggetto per paralisi respiratoria.
CLASSIFICAZIONE
Aldilà della classificazione operata sulla base della sintomatologia clinica (vedi dopo) in una forma di miastenia focale, generalizzata o fulminante, la classificazione più recente considera la relazione con il timoma.
Per ciascuna delle te forme cliniche, esiste una ulteriore classificazione in sottogruppi: si parla quindi di MG associata o non associata al timoma. Per la forma generalizzata, oltre ai sottogruppi associati e non associati al timoma, si considera un sottogruppo di soggetti sieronegativi per gli anticorpi e, nel gatto, un sottogruppo specifico per i soggetti in terapia con tireostatici (farmaci tiourilenici come il metimazolo e il carbimazolo).
DIAGNOSI
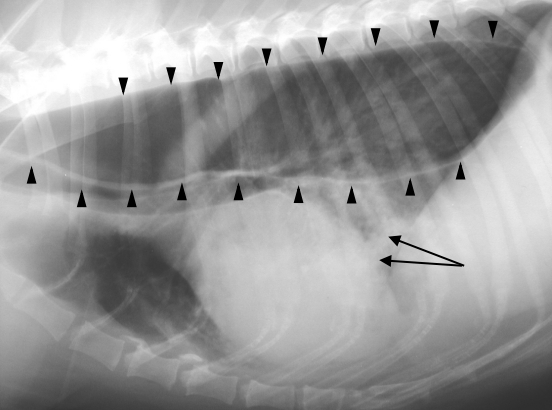
La diagnosi di miastenia gravis acquisita è multifattoriale e si compone di diverse procedure diagnostiche. Oltre al corredo sintomatologico, caratterizzato da intolleranza all’esercizio sovente associata a megaesofago, la diagnosi viene avvalorata da alcuni esami specifici. L’approccio ad un paziente con sospetta MG deve comprendere l’esecuzione di esami radiografici per evidenziare il megaesofago e identificare eventuali broncopolmoniti o neoplasie responsabili di sindromi paraneoplastiche (Figura 1).
Figura 1: Radiografia del torace di un cane affetto da MG. Le teste di freccia delimitano il megaesofago. Le frecce nere indicano invece una area di radioopacità compatibile con una polmonite "ab ingestis".
Il profilo ematobiochimico dei soggetti affetti da MG, in assenza di complicanze, è del tutto normale. La diagnosi definitiva si ottiene dimostrando la presenza di anticorpi sierici diretti contro i recettori per l’acetilcolina (Ab anti-AChR) tramite metodica radio-immunoenzimatica. Una concentrazione di anticorpi contro il recettore per l’ACh superiore a 0,30 nmol/L è positiva per MG acquisita nel gatto, mentre se è maggiore di 0,6 nmol/L indica un test positivo nel cane.
I falsi positivi sono estremamente rari; un risultato positivo del test è quindi considerato di conferma per la diagnosi di MG acquisita. Il test viene eseguito su un campione di siero.
Siccome i risultati del test di norma richiedono un certo lasso di tempo, una ulteriore conferma del sospetto diagnostico nel cane si può avere utilizzando il test con la neostigmina. Questi farmaci, entrambi ad azione anticolinesterasica, producono una immediata ma breve scomparsa dei segni clinici.
Il test con la neostigmina si esegue nel cane iniettando 40-50 µg/kg per via IM or 20 µg/kg per via EV. Considerata la maggior lunghezza di azione della neostigmina, è opportuno pretrattare il paziente con atropina (0.02–0.04 mg/kg) o glicopirrolato (0.01–0.02 mg/kg. il test è considerato positivo se l’animale, dopo l’inoculazione, manifesta un repentino aumento della forza, una risoluzione dell’astenia muscolare che però tende a ricomparire già dopo pochi minuti.
Uno studio recente (2021) ha dimostrato che il test con la neostigmina è risultato conforme alla diagnosi nell’81% dei casi. Va comunque ribadito che il test può dare falsi negativi o risultare positivo in presenza di altre malattie, come la polimiosite.
Infine, nell’ambito di una valutazione elettrodiagnostica, la miastenia gravis può essere dimostrata dai risultati del test della stimolazione nervosa ripetuta. Il test consiste nella stimolazione ripetuta a frequenza non superiore a 5 Hz del muscolo e nella registrazione del decremento del potenziale di azione muscolare composto (compound Muscle Action Potential – CMAP) oltre il 10% rispetto alla stimolazione iniziale. Questo test può essere estremamente utile nella diagnosi della MG focale o in situazioni clinicamente non chiare.
TERAPIA E PROGNOSI
La terapia di un soggetto affetto da Miastenia gravis autoimmune prevede in primis una serie di attenzioni volte ad evitare lo sviluppo di una broncopolmonite ab ingestis. Molti soggetti beneficiano di una posizione eretta durante l’assunzione del cibo in posizione eretta. Allo scopo, esistono diverse modalità, tra cui l’adattamento di piccoli tavolini o fare mangiare il cane sulle scale. L’irritazione esofagea, legata al megaesofago, può essere prevenuta con omeprazolo, somministrato una volta al giorno per bocca alla dose di 0,7/1 mg/kg.
Accanto alla terapia di sostegno è fondamentale la somministrazione di farmaci anticolinesterasici a lunga azione, come la piridostigmina bromuro (sciroppo o compresse: 0,5-3 mg/kg ogni 8-12 ore per os, cominciando ai dosaggi inferiori e aumentando gradualmente). In alternativa, è descritta la terapia associata di piridostigmina con dosi immunosoppressive a scalare di corticosteroidi. Questa scelta è poco utilizzata nel cane, mentre la terapia con soli corticosteroidi è la prima scelta nel gatto, estremamente sensibile agli effetti degli anticolinesterasici.
Nella MG gravis non associata al timoma, non è infrequente che sia il cane che il gatto vadano in remissione, soprattutto per quanto concerne i segni a carico dei muscoli appendicolari. La remissione clinica è definita come la risoluzione dei segni clinici, mentre la remissione immunologica è definita come la risoluzione dei segni clinici in concomitanza con la discontinuazione del trattamento e la normalizzazione della concentrazione sierica degli anticorpi anti-AChR. Il monitoraggio dell’andamento immunologico può essere effettuato con il dosaggio seriale degli anticorpi anti-recettori per l’ACh, effettuato ogni 6-8 settimane.
In un recente studio del 2021 su 94 cani, la remissione clinica (intesa dagli autori come assenza di segni clinici ≥4 settimane dopo la cessazione del trattamento) è stata osservata in 29 soggetti (31%), una risposta clinica (intesa come assenza di segni clinici durante il trattamento in 14 (15%) cani, il miglioramento clinico (inteso come riduzione dell’intensità della sintomatologia durante il trattamento) in 24 cani (26%), e nessun miglioramento clinico in 27 cani (29%).
La prognosi della MG deve essere considerata riservata ed è condizionata principalmente complicanza della broncopolmonite ab ingestis, complicazione più frequente e grave associata al megaesofago. La mortalità ad un anno di cani affetti da MG acquisita è riportata tra il 40 e il 60% dei soggetti.
La remissione immunitaria è frequente nel cane con MG focale e generalizzata non associata al timoma. Il trattamento può essere pertanto discontinuato e la prognosi nel lungo periodo può essere molto buona, fatto salvo quanto precedentemente detto in merito alla complicanza della BP ab ingestis.
È pertanto opportuno, in presenza anche di uno sfumato sospetto di complicanza broncopolmonare, intervenire in modo repentino con antibiotici ad ampio spettro. Nel gatto, forse per la minor incidenza del megaesofago, la prognosi della MG non associata al timoma è considerata migliore.
Bibliografia:
Shelton GD. Myasthenia gravis and disorders of neuromuscular transmission. Vet Clin North Am Small Anim Pract. 2002;32(1):189-206.
Dickinson PJ, LeCouteur RA. Feline neuromuscular disorder. Vet Clin North Am Small Anim Pract. 2004;34(6):1307-1359.
Shelton GD, Schule A, Kass PH. Risk factors for acquired myasthenia gravis in dogs: 1154 cases (1991-1995). J Am Vet Med Assoc. 1997; 211(1):1428-1431.
Shelton GD. Myasthenia gravis and congenital myasthenic syndromesin dogs and cats: a history and mini-review. Neuromuscul Disord. 2016;26(6):331-334.
Mignan T, Targett M, Lowrie M. Classification of myasthenia gravis and congenital myasthenic syndromes in dogs and cats. J Vet Intern Med. 2020 Sep;34(5):1707-1717. doi: 10.1111/jvim.15855. Epub 2020 Jul 15.
Forgash JT, Chang YM, Mittelman NS, Petesch S, Benedicenti L, Galban E, Hammond JJ, Glass EN, Barker JR, Shelton GD, Luo J, Garden OA. Clinical features and outcome of acquired myasthenia gravis in 94 dogs.J Vet Intern Med. 2021 Sep;35(5):2315-2326. doi: 10.1111/jvim.16223. Epub 2021 Jul 31.
Cridge H, Little A, José-López R, Pancotto T, Michaels JR, Menchetti M, Suñol A, Lipsitz D, Beasley MJ. The clinical utility of neostigmine administration in the diagnosis of acquired myasthenia gravis. J Vet Emerg Crit Care (San Antonio). 2021 Sep;31(5):647-655. doi: 10.1111/vec.13097. Epub 2021 Jul 29.
Gomes SA, Van Ham L, Van Ham A, Ives EJ, Vanhaesebrouck A. Canine Nonstructural Megaesophagus as a Clinical Sign of Potential Neurological Disease: 99 Cases. J Am Anim Hosp Assoc. 2020 Jan/Feb;56(1):7-16. doi: 10.5326/JAAHA-MS-6955. Epub 2019 Nov 12.
Gualtiero Gandini, Med. Vet. EBVS European Specialist in Veterinary Neurology (Dipl. ECVN); Esperto in Neurologia di MYLAV
Commenti
- Nessun commento trovato






Lascia i tuoi commenti
Login per inviare un commento
Posta commento come visitatore